Silyl Radical-Mediated Metallaphotoredox Cross-Electrophile Coupling
P. Zhang, C. C. Le, D. W. C. MacMillan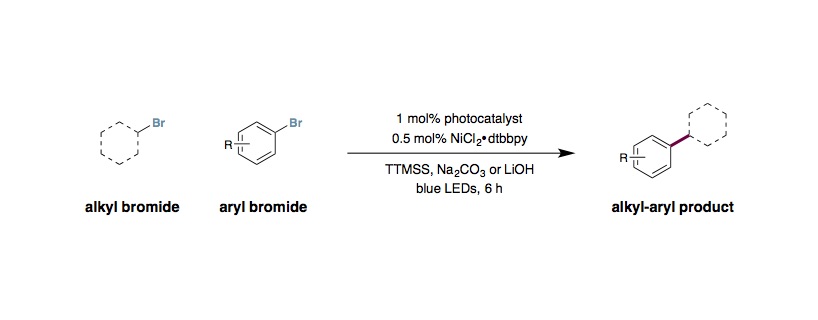
Light source:
34 or 40 W Blue LED lamp. Three 8 mL vials per lamp. Reaction time can usually be cut if you increase the number of lamps (6 hours for one lamp, 2 hours for two lamps).
General procedure:
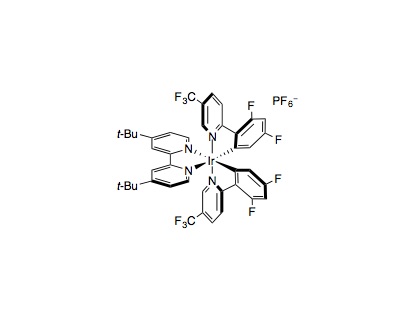
To an 8 mL vial, equipped with a stir bar, was added photocatalyst Ir[dF(CF3)ppy]2(dtbbpy)PF6 (2.5 μmol, 1 mol%), methyl 4-bromo benzoate (0.25 mmol, 1 equiv.), alkyl halide (0.375 mmol, 1.5 equiv.), tris(trimethylsilyl)silane (0.25 mmol, 1 equiv.), and anhydrous lithium hydroxide (0.5 mmol, 2 equiv.). The vial was sealed and placed under nitrogen before 2 mL of dimethoxyethane was added. To a separate vial was added NiCl2·glyme (0.013 mmol, 5 mol%) and 4,4´-di-tert-butyl-2,2´-dipyridine (0.013 mmol, 5 mol%). The catalyst vial was sealed, purged with nitrogen, and 1 mL of DME was then added. The precatalyst solution was sonicated or stirred for 5 minutes, after which, 0.1 mL of the solution (1.25 μmol, 0.5 mol% catalyst) was syringed into the reaction vessel. The reaction solution was then degassed by sparging with nitrogen while stirring for 10 minutes before sealing with Parafilm. The reaction was stirred and irradiated with a 34 W blue LED lamp (7 cm away, with cooling fan to keep the reaction temperature at 25 °C) for 6 hours. The reaction was quenched by exposure to air. Analysis of the crude reaction mixture can be performed by addition of mesitylene (0.250 mmol, 1.0 equiv.) as internal standard for 1H NMR. For isolation, dilute with DCM and wash with NaHCO3 (aq). Extract the aqueous layer with up to 3 DCM washes, and dry the combined organic layers with Na2SO4. Evaporation of the solvent and subsequent flash column chromatography provide the isolated product.
Tips and tricks:
- Anhydrous lithium hydroxide was obtained by heating LiOH-H2O to 100 °C at <100 mtorr.
- For reactions with heteroaryl bromides, the final product is often in equilibrium with the protonated form, which broadens the peaks in the 1H NMR. Before analysis of the reaction mixture, dilute with 2 mL of sat. NaHCO3 and 1 mL of water and extract with 4:1 DCM:iPrOH mixture (3 x 2 mL).
- For non-heteroarene products, the crude reaction mixture can be concentrated directly, with no need for workup.
- If solvent-coupled product is observed in DME, MeCN or dioxane can be used as alternative solvents.
- If necessary, dioxane (~ equal efficiency), MeCN (~ 50% efficiency), and DMA (generally low yields) can be explored in place of DME. A ratio of DMA:Toluene is also effective.
- LiOH is the best base for heteroaryl bromides, while Na2CO3 can be used for non-heteroaryl substrates.
- Homogenous conditions utilizing 2,6-lutidine as a base are comparable in efficiency and ideal for flow. For heterocycles, 5 equiv of 2,6-lutidine is recommended to achieve similar yields as anhydrous LiOH.
- Two equivalents of base are generally needed, but some substrates can require additional equivalents.
- 1.5 equivalents of alkyl bromide are not always required- often 1 equivalent can provide the product in comparable efficiency. Typically, increasing to 2 equivalents of alkyl bromide can provide higher yields.
- Tertiary alkyl bromides require increased reaction time (48-72 hours), 1.5 or mroe equivalents of silane, and higher Ni loadings (10 mol%).
- Increasing silane equivalents can also increase the yield for low yielding substrates.
- For non-polar substrates, reverse phase chromatography is recommended for purification.
Every nickel source we evaluated provides product, except for Ni(OH)2. Competent nickel sources include NiBr2·glyme, Ni(BF4)2, Ni(OAc)2, NiCl2·6H2O, NiBr2·3H2O, Ni(acac)2, and Ni(stearate)2.