Arylation of sp3 C-H Bonds via Metallaphotoredox/HAT Catalysis
M. H. Shaw, V. W. Shurtleff, J. A, Terrett, J. D. Cuthbertson, D. W. C. MacMillan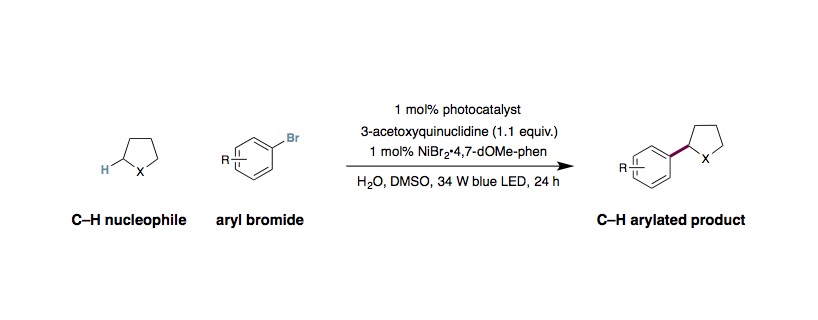
Light source:
34 W blue LED, two 8 mL vials can be placed in front of each lamp.
General procedure:
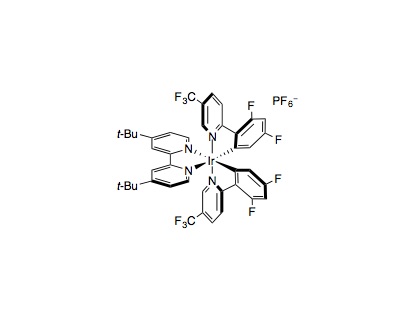
To an 8 mL vial equipped with a magnetic stir bar were added Ir[dF(CF3)ppy]2(dtbbpy)PF6 (0.005 mmol, 1 mol%), aryl bromide (0.50 mmol, 1 equiv.), the Csp3–H nucleophile (1.00 mmol, 2 equiv.) and anhydrous DMSO (1.0 mL). Next, NiBr2•3H2O (0.005 mmol, 1 mol%) and 4,7-dOMe-1,10-phenanthroline (0.005 mmol, 1 mol%) were added as a 0.5 M solution in anhydrous DMSO (1 mL, a stock solution of Ni-catalyst and ligand in DMSO was sonicated for 10 minutes prior to addition). Water (20.0 mmol, 40 equiv.) and 3-acetoxyquinuclidine (0.55 mmol, 1.1 equiv.) were added to the vial via microliter syringe. The vial was sealed and the reaction mixture was degassed via two sequences of freeze-pump-backfill-thaw. The reaction was sealed with parafilm and placed 6 cm away from a 34 W blue LED lamp and stirred with cooling by fan (reaction time varies with substrate, generally complete within 24 h). The reaction mixture was removed from the light and diluted with EtOAc (15 mL) and water (20 mL). The layers were separated and the aqueous portion was further extracted with EtOAc (2×15 mL). The organic extracts were combined, dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography to afford the cross-coupled product.
Tips and tricks:
- Degassing via freeze-pump-backfill-thaw cycles was carried out as follows: The reaction mixture, in a vial equipped with a N2 inlet line, was submerged in a dry ice/acetone bath for 2–5 minutes, until the mixture was frozen. The vial was evacuated for 5 minutes, then backfilled with N2 and allowed to warm to ambient temperature. The mixture was then stirred for ~10 sec and the process was repeated. Approximately the same yields were obtained when the reaction mixture was sparged with N2 for 15 minutes instead. When using this method, the water was sparged separately and added after degassing the reaction mixture.
- For optimization of a particular substrate, the key reaction parameters which had the largest influence on the yield were equivalents of water, reaction concentration (often water loading requires adjusting with concentration) and solvent (DMSO and MeCN were the best solvents evaluated).
- Pyridine HCl salts can be utilized by increasing the amount of 3-acetoxyquinuclidine to 2.1 equivalents.
- Generally, the reaction works best when aryl bromides are employed; however electron-deficient aryl chlorides can also be competent reaction partners. In particular, for pyridines bearing electron-withdrawing substituents (i.e. F, CF3) the aryl chloride often performed better than the aryl bromide. In these cases, higher yields were achieved when water was excluded from the reaction conditions.
- If protodehalogenation or phenol byproducts are observed, try lowering the loading of Ni catalyst.
- If phenol byproducts are observed, try adjusting the water loading. It may seem counterintuitive but sometimes increasing the water loading significantly reduced formation of phenol byproducts.
- For difficult C–H nucleophile substrates, addition of 3-acetoxyquinuclidine in two batches (0.55 equiv. at start of reaction and 0.55 equiv. after 6-10 h) sometimes helped to improve reaction efficiency.
- Modified reaction conditions were required for 2-halopyridines in which quinuclidine (1.1 equiv.) was used in place of 3-acetoxyquinuclidine with MeCN (0.25 M) as solvent and increased water loading (100 equiv.).
- The reaction can be scaled up by conducting the cross-coupling in a 40 mL vial. In these cases, the reaction vial was placed between two 34 W blue LED lamps (approx. 6 cm from each).