Metallaphotoredox Decarboxylative Alkylation
C. P. Johnston, R. T. Smith, S. Allmendinger, D. W. C. MacMillan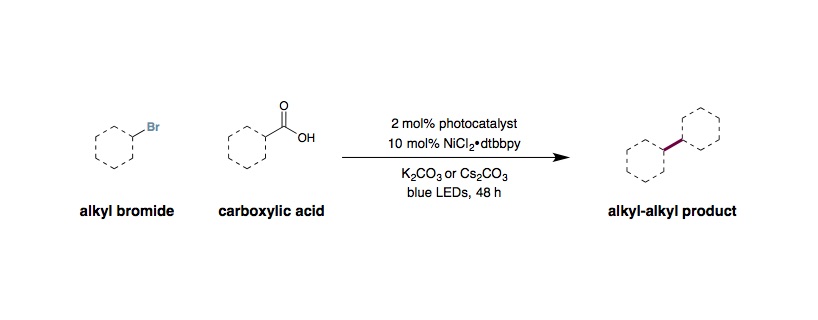
Light source:
Kessil Blue LED lamps. For 40 mL vials, use 2 lights per reaction. For 8 mL vials, use 1 light per reaction.
General procedure:
For a-amino carboxylic acids:
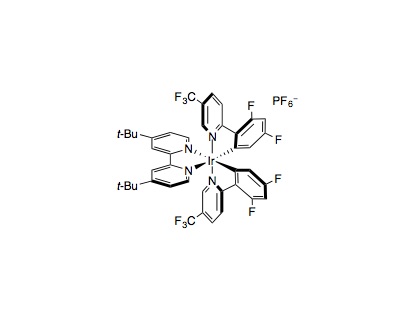
An oven dried 8 mL vial equipped with a Teflon septum and magnetic stir bar (1/2” x 1/8”, Fisher 1451393) was charged with Ir[dF(CF3)ppy]2(dtbbpy)PF6 (0.01 mmol, 2 mol%), NiCl2⋅glyme (0.05 mmol, 10 mol%), 4,4¢-dimethoxy-2,2¢-bipyridine (0.05 mmol, 10 mol%), Boc-Pro-OH (0.75 mmol, 1.5 equiv.), K2CO3 (1 mmol, 2 equiv.), and MeCN (5 mL, 0.1 M). The reaction mixture was degassed by bubbling nitrogen stream for 15 min. Water (10 mmol, 20 equiv.) and the appropriate alkyl halide (0.5 mmol, 1 equiv.) were then added. The reaction mixture was then stirred and irradiated with two 34 W blue LEDs (vials approximately 6 cm away from the light source) with a fan placed above for cooling. After 24–48 h (primary bromides are usually done within 24 hrs or earlier), the reaction mixture was diluted with EtOAc, filtered (through cotton, to remove the inorganic base), and concentrated in vacuo. Purification of the crude product by flash chromatography on silica gel afforded the desired product.
For acyclic a-amino carboxylic acids:
Follow above procedure, but dilute reaction 2-fold (possibly requiring 40 mL vial, cross-shape stir bar) and use Cs2CO3.
For aliphatic carboxylic acids:
An oven dried 40 mL vial equipped with a Teflon septum and cross-shape magnetic stir bar was charged with Ir[dF(F)ppy]2(dtbbpy)PF6 (0.01 mmol, 2 mol%), NiCl2⋅glyme (0.05 mmol, 10 mol%), 4,4¢-di-tert-butyl-2,2¢-bipyridine (0.05 mmol, 10 mol%), acid (1.0 mmol, 2 equiv.), Cs2CO3 (1.5 mmol, 3 equiv.), and 1:1 EtOAc/MeCN (22 mL, 0.02 M). The reaction mixture was degassed by bubbling nitrogen stream for 15 min. Water (7.5 mmol, 15 equiv.) and bromide (0.5 mmol, 1.0 equiv.) were then added. The reaction mixture was then stirred and irradiated with two 34 W blue LEDs (vials approximately 6 cm away from the light source) in a water bath with a fan placed above for cooling. After 24 h, an additional portion of Ir[dF(F)ppy]2(dtbbpy)PF6 (0.01 mmol, 2 mol%) was added in a minimal amount of MeCN (degassed). After a further 24 h of irradiation, the reaction mixture was diluted with EtOAc, filtered (through cotton, to remove the inorganic base), and concentrated in vacuo. Purify by silica chromatography.
Second addition of photocatalyst was necessary to drive the reaction to full completion so that no bromide was remaining (left over bromide complicated product purification for our specific substrates). The water bath ensures the reaction temperature is kept near ambient temperature and can reduce the amount of ester byproduct observed with certain substrates.
For running the decarboxylative methylation:
The procedure for a-amino carboxylic acids may be followed using a pre-prepared solution of bromomethane, with the only difference being that the acid should be used as the limiting reagent (typically, slightly more than 1 equivalent of MeBr was added in solution, but more can also be used). We prepared the stock solution in MeCN, but if a solution in THF, EtOAc, or acetone is available, these solvents are known to be tolerated. Vast excess of MeBr promotes esterification, but the reaction will still work (if a vast excess is desired, varying water equivalents, base, and temperature can help to overcome this issue). If desired, methyl tosylate (MeOTs) may instead be used for this coupling in the presence of excess cesium bromide (5-7 equiv.). Esterification is an issue under these conditions, and some optimization may be required (again: water, base, and temperature are probably the best things to check first).
Tips and tricks:
- The amount of water used can be important for some acids. This will also depend on the base used. Potassium and cesium carbonate tend to need more water than organic bases (such as tetramethylguanidine). It might be worth investigating different quantities of water.
- Cesium carbonate is a more generally successful base, but in some cases potassium carbonate is slightly better. Tetramethylguanidine (which ensures a homogeneous solution amenable to flow conditions) has been successful with proline, but it has not been tried with other acids at present.
- Dilution can sometimes be beneficial, and in some cases adding EtOAc will help. This is useful if esterification is a major byproduct or if you are having issues with reductive bromide dimerization (a problem with primary bromides). Alternative non-polar co-solvents, such as dioxane and tetrahydropyran, can also be useful.
- Ir[dF(CF3)ppy]2(dtbbpy)PF6 and Ir[dF(F)ppy]2(dtbbpy)PF6 have both proven successful in this transformation. If a low yield is observed with one photocatalyst try the other one or try additional quantities of photocatalyst if starting materials are remaining.
- Difficult substrates. Some aliphatic acids may prove difficult and require optimization due to destruction of the photocatalyst by the alkyl radical. Coupling of secondary bromides with secondary acids is difficult except in the case of proline. Coupling of a primary component (either acid or bromide) with a secondary one is generally more successful. This is a common problem with Ni catalyzed alkyl-alkyl couplings.
- Sometimes higher yields are observed when using a 40 ml vial with a large cross stir bar (better vortex, light penetration, etc.).
- Sometimes reactions tend to form inconsistent suspensions while degassing, depending on stir rate, time span between mixing and irradiating etc, leading to reproducibility issues. In these cases, the reactions should be sonicated (~1-2 min) after sparging and before irradiation to ensure a reproducible “homogeneous suspension.”
If desired, it is often possible to invert the stoichiometry of the reaction (i.e., use excess bromide) when using primary electrophiles. This may affect the yield positively or negatively, but the reaction should still work.