Metallaphotoredox Double Decarboxylative Arylation
X. Zhang, D. W. C. MacMillan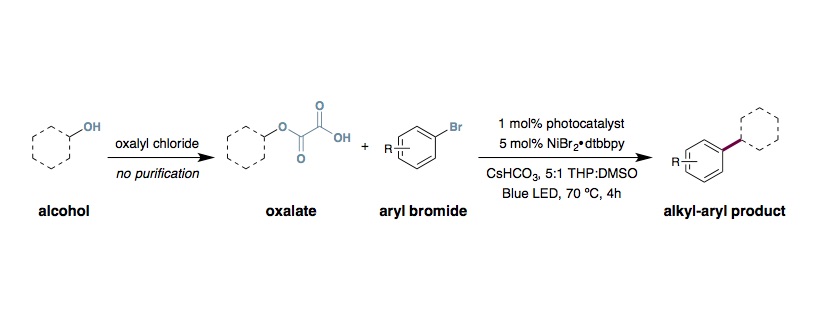
Light source:
Kessil Blue LED lamp. 2 or 4 lamps for a set of 2 40 mL vials.
General procedure:
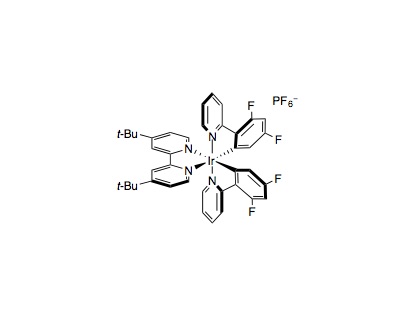
To an oven-dried 40 mL vial equipped with a stir bar was added photocatalyst Ir[dFppy]2(dtbbpy)PF6 (4.0 mg, 4.0 µmol, 0.01 equiv.), methyl 4-bromobenzoate (86 mg, 0.40 mmol, 1.0 equiv.), 2-((1-(tert-butoxycarbonyl)piperidin-4-yl)oxy)-2-oxoacetic acid (142 mg, 0.52 mmol, 1.3 equiv.) and anhydrous cesium hydrogencarbonate (116 mg, 0.60 mmol, 1.5 equiv.). The vial was sealed and placed under nitrogen before THP (6 mL) and DMSO (2 mL) was added. To a separate vial was added NiBr2•glyme (6.2 mg, 20 µmol, 0.05 equiv.) and 4,4′-di-tert-butyl-2,2′-bipyridine (5.4 mg, 20 µmol, 0.05 equiv.). The pre-catalyst vial was sealed, purged with nitrogen, dissolved in THP (4 mL) and then sonicated until it became homogeneous. Subsequently, the catalyst was syringed into the reaction vessel and the solution was degassed by sparging with nitrogen while stirring for 15 minutes before sealing with parafilm. The reaction mixture was preheated to 70 °C. Then the reaction was stirred and irradiated using 34 W blue LED lamps (1 cm away without cooling fan to heated the reaction to approximately 70 °C, see figure S1) for 4 hours. The reaction mixture was removed from the light, cooled to ambient temperature, diluted with water and EtOAc, and the aqueous layer was extracted with three portions of EtOAc. The combined organic layers were washed with brine, dried over sodium sulfate, filtered, and concentrated. The residue was purified by flash chromatography on silica gel to afford the desired alkyl-aryl product.
Tips and tricks:
- It is critical to preheat the reaction to 70 °C. The elevated temperature is required to achieve efficient double decarboxylation and give alkyl-aryl product, as opposed to aryl ester product. If a substrate is giving large amounts of ester, increasing the reaction temperature (using the lights) to 80 or 85 °C can help.
- Dioxane is comparable to THP in most reactions. Playing with the DMSO ratio in the solvent mixture can be helpful to deal with protodehalogenation (more DMSO) or oxalate hydrolysis (less DMSO). Adjust the ratio as needed for whatever problems you find with each substrate.
- The reaction can be concentrated to 0.1 M and still produce useful yields of product (30-40%).
- Nickel loading can be adjusted to help deal with protodecarboxylation (more Ni, for more efficient radical capture) and protodehalogenation (less Ni). You need to strike a balance.
- Cs bases seem to be essential. The second base base is cesium carbonate.
- Oxalates can be prepared according the following procedure and used without purification:
A round-bottom flask was charged with alcohol (1.0 equiv) followed by the addition of 3:1 Et2O/dichloromethane (0.15 M). The solution was cooled to 0 °C. Next, oxalyl chloride (2.0 equiv) was added dropwise. The homogeneous reaction mixture was allowed to warm to ambient temperature and stirred for 18 h. The reaction was then cooled to 0 °C and quenched by slow addition of H2O (30% v/v). After stirring for 1 h at ambient temperature, the resulting mixture was then transferred to a separatory funnel, and the aqueous layer mixture was extracted with three portions of Et2O. The combined organic layers were washed with brine, dried over sodium sulfate, filtered, and concentrated under reduced pressure. It is important to rigorously dry the oxalates. If hi-vac does not dry them sufficiently, azeotrope with toluene a few times, and then hi-vac.